欧州医療機器規則 (MDR)2017/745

|
2020年4月23日、EU Council (EU理事会)は、MDRの1年延期の提案を採択したことを発表しました。翌4月24日、EU官報(オフィシャルジャーナル)が発行されたことで、MDRの適用開始日を1年後に延期する措置が正式に発効されました。 この決定は、主要な医療機器の市場での不足や遅延を防ぐために欧州委員会が作成した提案に基づいて行われ、欧州議会及び欧州理事会は医療機器規制の適用日を1年延期(2021年5月26日)することを決定したものです。 御参考:EU官報(オフィシャルジャーナル) https://eur-lex.europa.eu/eli/reg/2020/561/oj |
|---|
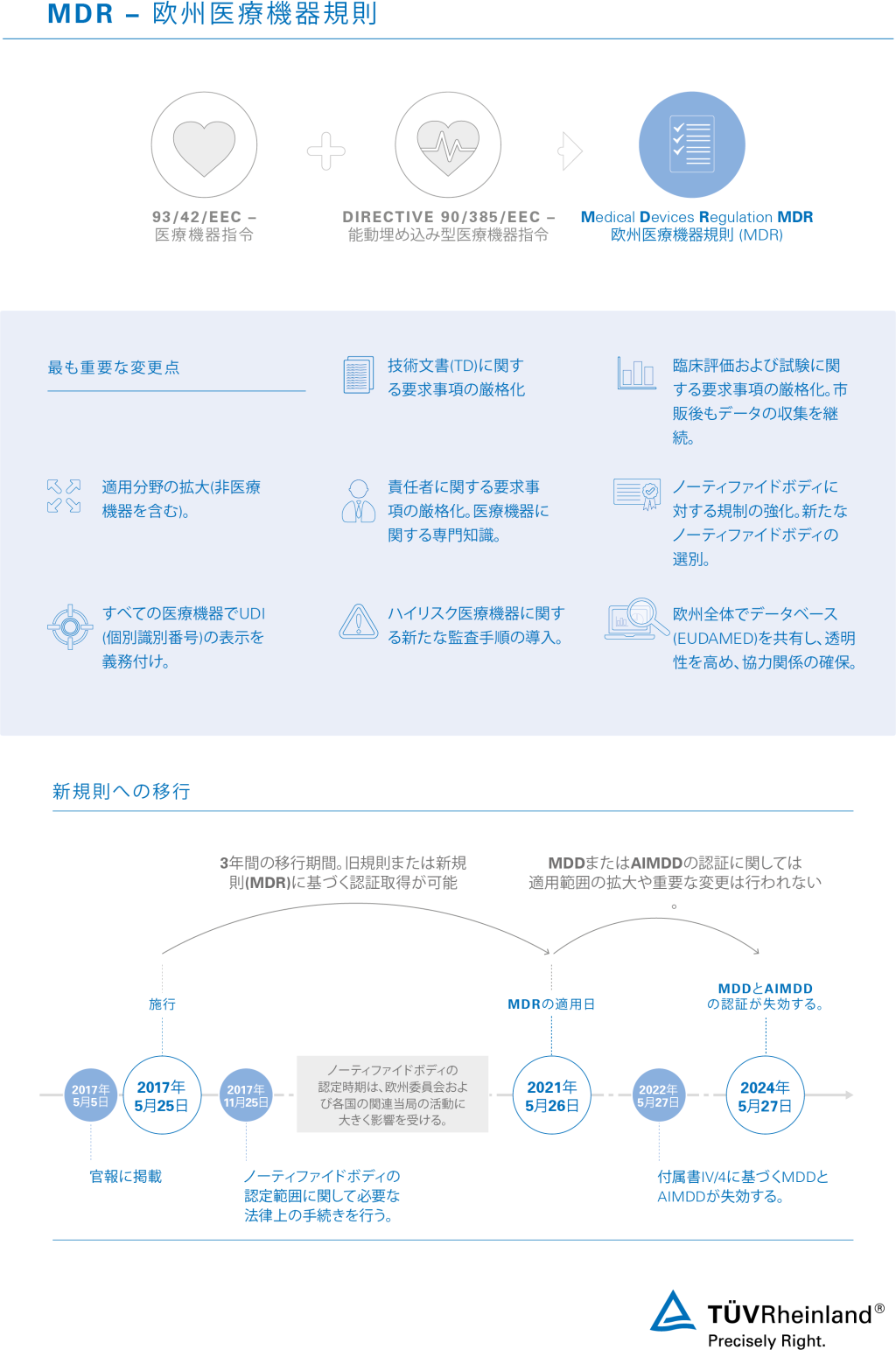
欧州医療機器規則 (MDR)に対する適合性評価を早期に開始するメリット

MDR監査を受けて欧州市場への参入を実現
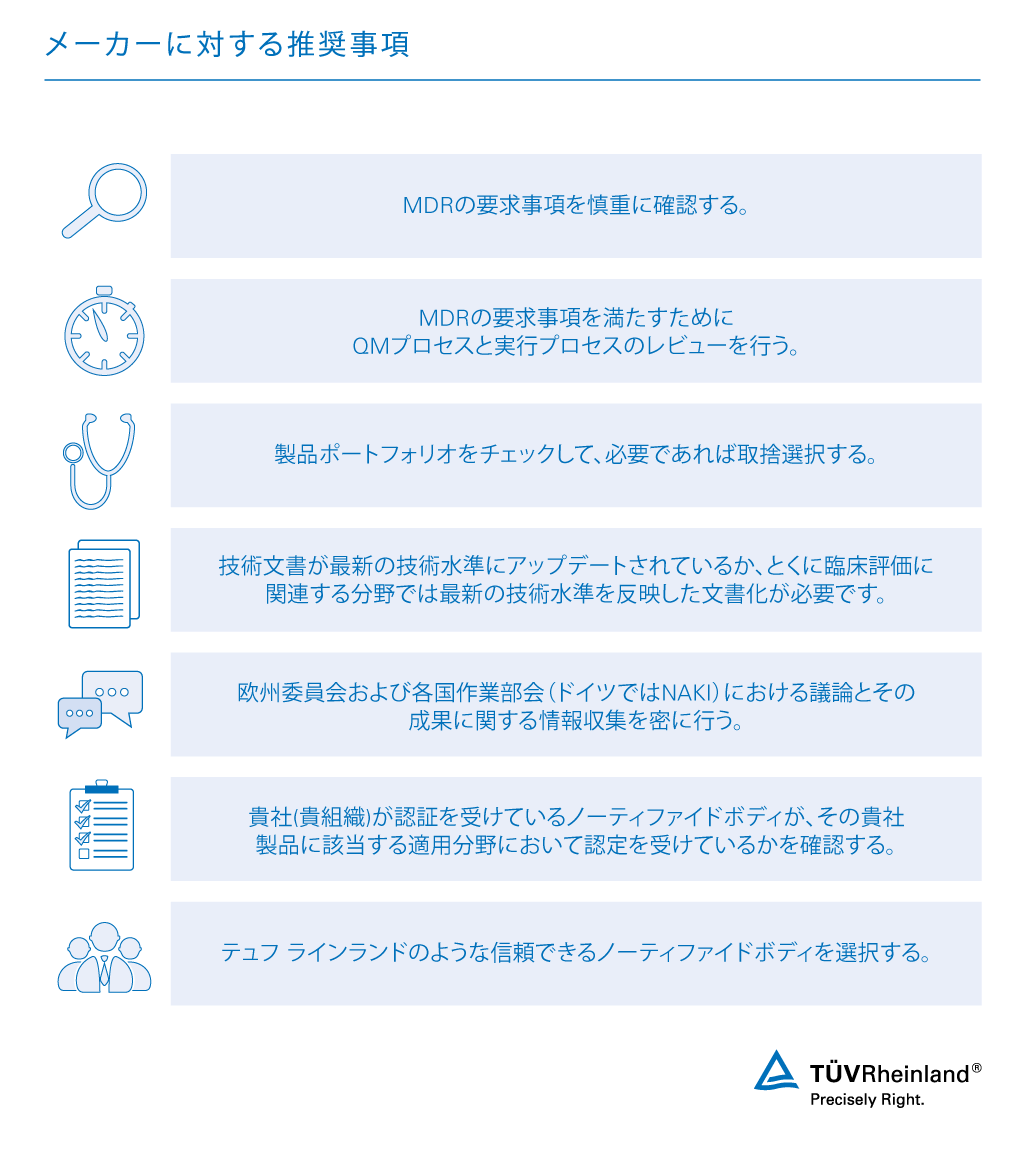
欧州で市販される医療機器を規制する従来の規則は、新規則のMDR 2017/745によって置き換えられます。このため製造業者は、新規則に基づく適合性の評価を行う必要があります。MDRの適合性評価を行えば、欧州市場で医療機器を販売するうえで必要となる認証を取得することができます。新規則に基づく認証を取得するには、製品が要求事項を満たしていなければなりません。
テュフ ラインランドでは、製品の適合性を確保するためのアドバイスや情報提供のほか、技術文書のレビューや監査も行っています。業界経験豊富な専門家およびグローバルなネットワークを生かして、 医療機器関連サービス をワンストップで提供しています。
医療機器の適合性評価とその他のサービス
テュフ ラインランドでは、MDR 2017/745への移行期間および移行スケジュールに関するサービスを提供しています。移行期間内に確実に問題を解決できるよう専門家がサポートを行い、欧州市場における医療機器の販売を継続可能にします。
信頼できるパートナーが医療機器指令の新規則への移行をサポート
テュフ ラインランドはグローバルなビジネスをサポートする試験・認証機関として、医療機器業界にワンストップで包括的なサービスを提供しています。新たに施行された欧州医療機器規則への移行をサポートするだけでなく、医療機器メーカー、サプライヤー、セールスオフィスにおけるQMシステムの監査から医療機器の試験まで、そのサービス内容は多岐に渡ります。時代のイノベーションに適用する認証機関として、無線通信、遠隔治療、医療アプリ、 サイバーセキュリティ、 個人情報保護など、デジタル化分野にも対応しています。
早めにMDRの適合性評価手続きを開始しましょう。ぜひお問い合わせください。
新欧州医療機器規則 (MDR 2017/745) に関するQ&A



英国マーケットへのサポート

テュフ ラインランドは、医療機器(体外診断用医療機器含む、以下、医療機器等)の英国への市場参入に必要な知識と経験を有する専門家が揃っています。テュフ ラインランド UKは、UKCAマーキングのApproved Bodyとして、英国医療機器規制2002 (SI 2002 No 618, as amended) (UK MDR 2002)に基づき、医療機器等の適合性評価および認証を行うことができます。
UKCA認証に関して/MDR移行期間における条件および注意点
- UKCA認証に関するQ&A
- 移行期間における条件および注意点
/tuv-rheinland-japan-official-blog-for-medical-image_core_1_x.jpg)
テュフ ラインランド ジャパン 公式ブログ
では、
・最新規制情報
・専門家の声
・お客様の事例
をブログにてご紹介しております。
最新情報を、ぜひ業務にお役立てください。
公式ブログは こちら
各国規制・認証検索サービス、国際規制情報
各国規制・認証検索では、現地試験の必要性や認証の有効期限など、各国の規制・認証の概要を英語で提供しています。国際規制情報では、世界の規制・国際規格に関する最新情報を英語で提供しています。



オンラインマガジン「tuv.communication」配信登録

月に一度、テュフ ラインランドの最新情報をまとめて、皆さまにオンラインマガジンとしてお届けしています。国際規制情報・新サービス・事例紹介・セミナー情報などをまとめて取得できます。ぜひこの機会にご登録ください。
配信コンテンツ
・業務に役立つ最新国際規制情報
・当社の新サービス
・事例紹介
・セミナーご案内
配信頻度
月1回


/tuv-rheinland-de19_p05_ivd09-lp_core_4_3.jpg)
/tuv-rheinland-medical-device-single-audit-program-mdsap-ad-106934074_core_4_3.jpg)
{kind=link}