Rozporządzenie UE – certyfikacja wyrobów medycznych – certyfikat MDR 2017/745

Przedłużono okres przejściowy MDR
16 lutego 2023 roku Parlament Europejski zatwierdził wniosek, który znacznie złagodzi wyzwania producentów wyrobów medycznych. Dzięki najnowszemu rozszerzeniu rozporządzenia w sprawie wyrobów medycznych UE 2017/745 mają oni możliwość ubiegania się o przedłużenie okresu przejściowego dla swoich produktów.
Nowe rozporządzenie ma wejść w życie już niebawem, po publikacji w Dzienniku Europejskim. Producenci będą mieli wtedy perspektywę następujących okresów przejściowych:
- 26 maja 2026 r. wyroby medyczne (wykonywane na zamówienie, klasa III)
- 31 grudnia 2027 r. wyroby medyczne (implanty wyższego ryzyka, nieobjęte wyłączeniem, klasa IIb i wyroby klasy III)
- 31 grudnia 2028 r. wyroby medyczne (niskiego ryzyka)
Należy zaznaczyć, że producenci muszą wykazać, że podjęli już kroki w celu dostosowania się do nowych przepisów, aby ubiegać się o przedłużenie terminu. Oferujemy wsparcie dla wszystkich producentów, którzy szukają profesjonalnej i wiarygodnej Jednostki Notyfikowanej, aby sprostać wyzwaniom stawianym przez MDR i IVDR. Skontaktuj się z nami!
|
Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2020/561 z dnia 23 kwietnia 2020 roku zmieniające rozporządzenie (UE) 2017/745 w sprawie wyrobów medycznych zostało opublikowane w dniu 24 kwietnia 2020 roku w Dzienniku Urzędowym Parlamentu Europejskiego! Głównym celem poprawki jest przesunięcie terminu stosowania z 26 maja 2020 roku na 26 maja 2021 roku wraz z tym przesunięciem przyjęto również inne terminy stosowania dodatkowych przepisów. Oficjalna publikacja rozporządzenia zmieniającego znajduje się tutaj. |
|---|
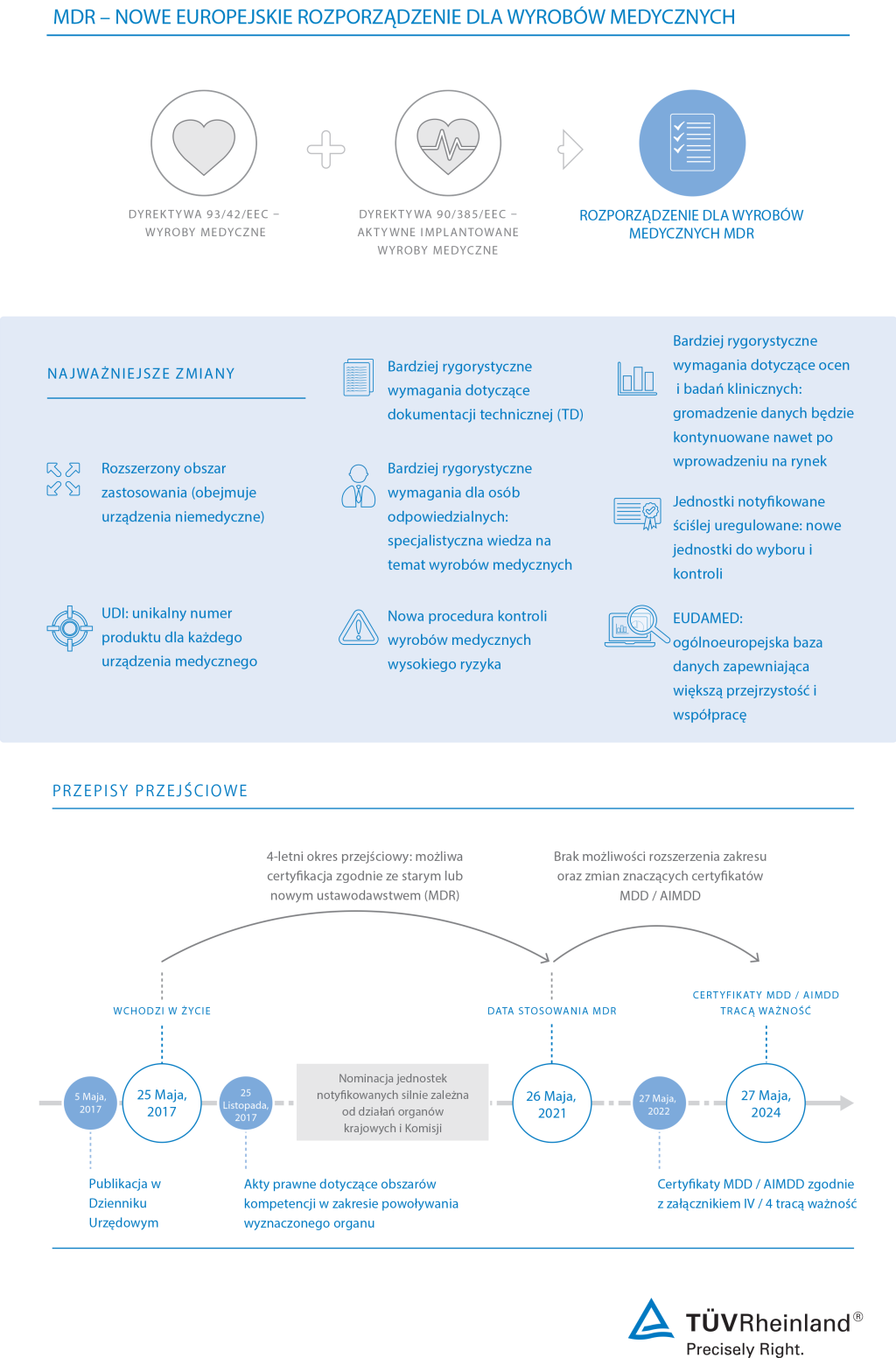
Szybki start w zakresie zgodności z nowym rozporządzeniem UE w sprawie wyrobów medycznych

Producenci wyrobów medycznych stoją w obliczu nowych wymagań zgodnie z MDR 2017/745, który wszedł w życie 25 maja 2017 r. Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2020/561 z dnia 23 kwietnia 2020 r. zmieniające rozporządzenie (UE) 2017/745 które opóźnia datę stosowania do 26 maja 2021 r. TÜV Rheinland docenia i popiera.
Środek ten daje producentom urządzeń medycznych możliwość pełnego skoncentrowania wszystkich możliwych zasobów na walce z pandemią COVID-19. Producenci urządzeń medycznych odgrywają ważną oraz wymagającą rolę. Niezwykle ważne jest, aby wyroby medyczne były zgodne i nadal były dostępne w UE, aby uniknąć niedoborów lub opóźnień niektórych wyrobów medycznych w tych wyjątkowych okolicznościach.
Poza tą sytuacją TÜV Rheinland chciałby zachęcić swoich klientów do utrzymywania z nami kontaktu w celu dalszego planowania działań w odniesieniu do wdrożenia rozporządzenia w sprawie wyrobów medycznych, które będzie stosowane od dnia 26 maja 2021 r.
Audyty MDR zapewniają dostęp do rynków UE
Dzięki zastąpieniu przez MDR 2017/745 istniejących przepisów dotyczących wyrobów medycznych w UE producenci będą wkrótce zobowiązani do ponownej oceny swoich wyrobów pod kątem zgodności. Procedura oceny zgodności MDR daje wytwórcom szansę na uzyskanie niezbędnej certyfikacji wymaganej do wprowadzenia produktów na rynek europejski. Dostęp do zgodności, warunek wstępny sukcesu regulacyjnego.
Jako element procedury oceny zgodności jednostka notyfikowana prowadzi wymagane przeglądy dokumentacji technicznej wyrobów oraz audyty. Jesteśmy wspierani przez globalną sieć doświadczonych w branży specjalistów i placówek oraz oferujemy kompleksową obsługę usług związanych z wyrobami medycznymi.
Ocena zgodności wyrobu medycznego i nie tylko
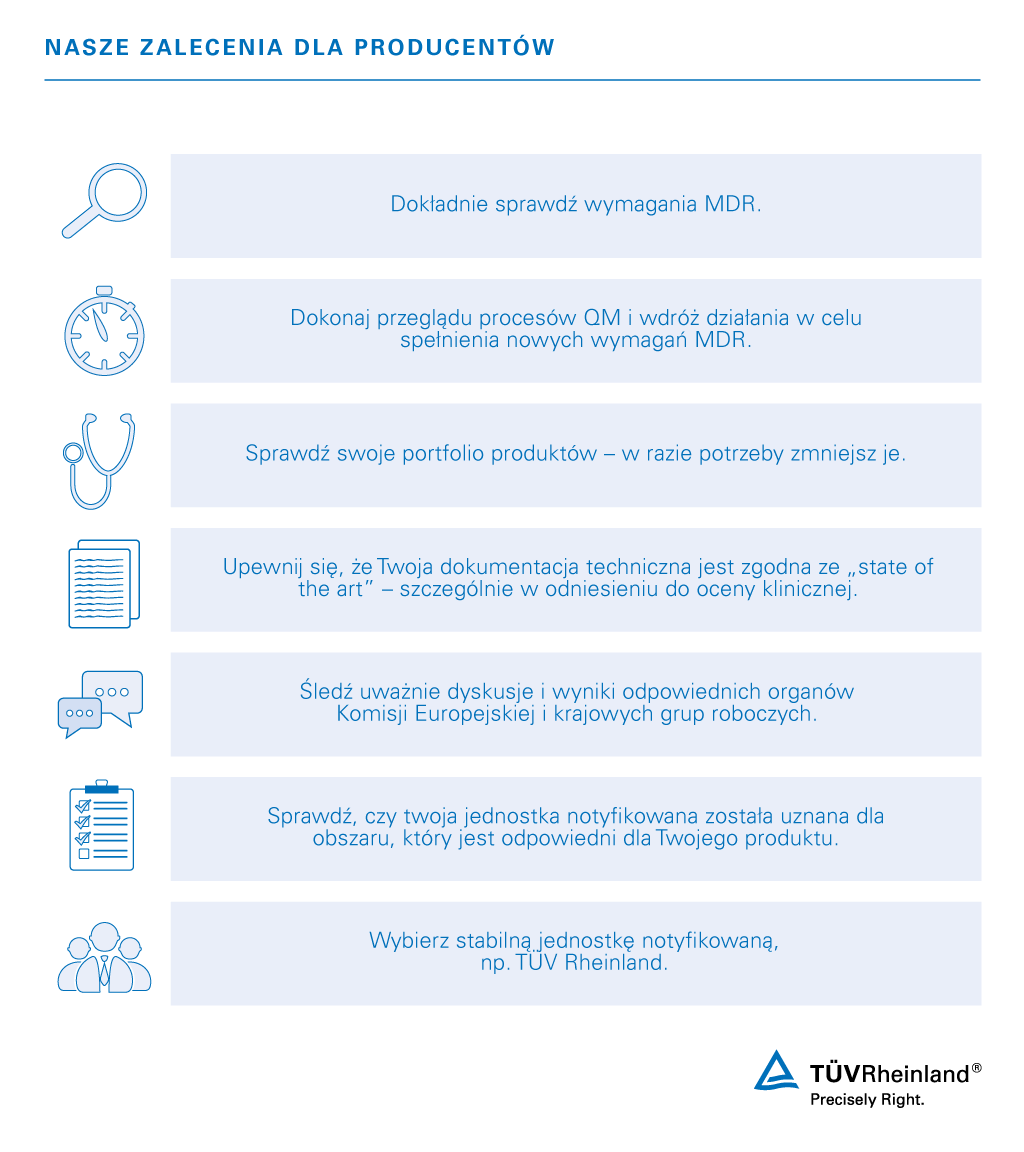
Obecnie nasze usługi koncentrują się na okresie przejściowym i harmonogramie zgodności z MDR 2017/745. Nasi eksperci mogą pomóc w dotrzymaniu terminów i rozwiązaniu wszystkich problemów związanych z utrzymaniem dostępu Twojego sprzętu medycznego do rynków europejskich.
Twój pewny partner w przeprowadzeniu zastąpienia dyrektywy dotyczącej wyrobów medycznych
Jako organizacja testująca i certyfikująca oraz specjalista w zakresie dostępu do rynków na całym świecie, oferujemy kompleksową obsługę branży urządzeń medycznych pod jednym dachem. Oprócz umożliwienia przejścia do zgodności z nowym europejskim Rozporządzeniem w Sprawie Wyrobów Medycznych, nasze usługi obejmują audyt systemu zarządzania jakością dla producentów, dostawców i biur sprzedaży urządzeń medycznych, a także badania urządzeń medycznych. Jesteśmy innowacyjni i przygotowani na przyszłe wyzwania z zakresu digitalizacji, takie jak połączenia bezprzewodowe, telemedycyna, aplikacje medyczne, bezpieczeństwo cybernetyczne, ochrona danych osobowych i inne.
Skontaktuj się z ekspertem, aby już zacząć spełniać wymogi zgodności z MDR.
Pytania i odpowiedzi dotyczące rozporządzenia UE w sprawie wyrobów medycznych MDR 2017/745
Więcej informacji do pobrania
| Deklaracja zainteresowania MDR | 80 KB | Pobierz |
Kontakt


/tuv-rheinland-medical-device-single-audit-program-mdsap-ad-106934074_core_4_3.jpg)
/tuv-rheinland-de19_p05_ivd09-lp_core_4_3.jpg)
{kind=link}