EU-Medizinprodukteverordnung MDR 2017/745

News: MDR-Übergangsfristen verlängert

Es wird erwartet, dass die neue Verordnung in Kürze durch Veröffentlichung im Europäischen Journal in Kraft tritt. Herstellern werden dann die folgenden Übergangsfristen in Aussicht gestellt:

Bitte beachten Sie, dass die Hersteller nachweisen müssen, dass sie bereits Schritte zur Umstellung auf die neuen Vorschriften unternommen haben, um die Verlängerung in Anspruch nehmen zu können. Wir bieten Unterstützung für alle Hersteller, die eine professionelle und zuverlässige Benannte Stelle suchen, um die Herausforderungen von MDR und IVDR zu meistern. Jetzt kontaktieren!
Vorsprung durch Umstellung auf die neue EU-Medizinprodukteverordnung

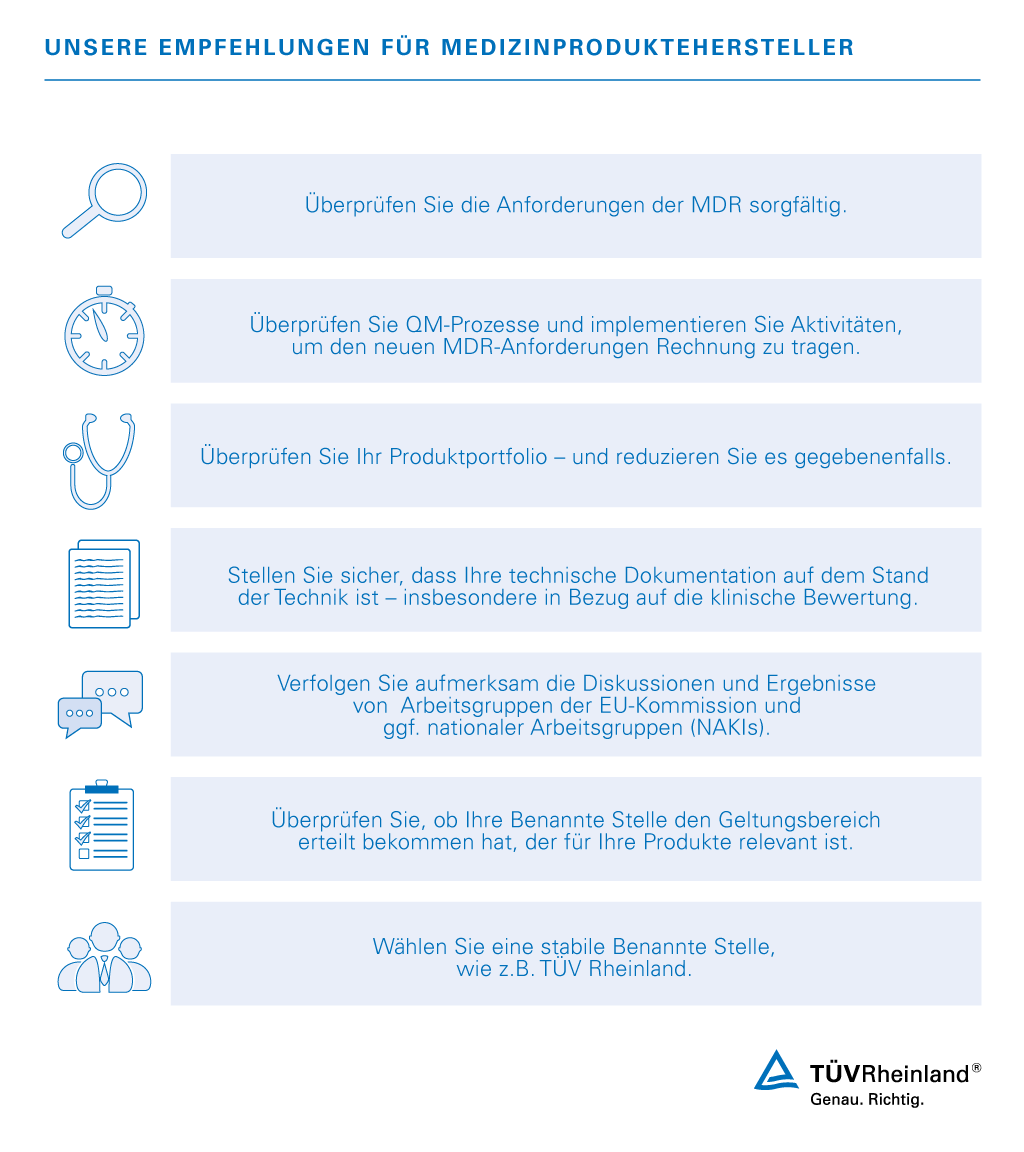
Hersteller von Medizinprodukten sehen sich durch die neue Medizinprodukteverordnung 2017/745 (Medical Device Regulation, MDR) neuen Anforderungen gegenüber. Die Verordnung trat am 25. Mai 2017 in Kraft.
Als etablierte Benannte Stelle haben wir unsere Expertise und Zuverlässigkeit bei der Beteiligung an Ihren Konformitätsbewertungsverfahren für die Richtlinien 93/42/EEC für Medizinprodukte (MDD) und 90/385/EEC für aktiv implantierbare Medizinprodukte (AIMDD) bereits unter Beweis gestellt. Beide Richtlinien werden nun durch die neue Verordnung ersetzt. Unsere Leistungsfähigkeit und Kompetenz sind in der Medizinbranche etabliert, und wir freuen uns darauf, unser Wissen mit unseren Kunden zu teilen.
Mit unserer Erfahrung und unserer Reputation als Benannte Stelle für Medizinprodukte sind wir Ihr perfekter Partner, um Sie beim Übergang zur neuen Verordnung zu unterstützen.
Erfahren Sie mehr über Details und den Zeitplan der MDR 2017/745 sowie darüber, welchen Einfluss sie auf Ihre Geschäfte hat.
Konformität zur neuen MDR sichert den Zugang zu den europäischen Märkten
Da die aktuell gültigen Richtlinien für Medizinprodukte in der EU durch die MDR 2017/745 ersetzt werden, müssen Hersteller ihre Produkte bald erneut auf Konformität überprüfen. Ein Konformitätsbewertungsverfahren nach MDR entspricht immer einer Neu-Zertifizierung und ist für das Inverkehrbringen von Medizinprodukten in Europa verpflichtend.
Wir stehen Ihnen als zuverlässiger Partner während der Umstellungsphase zur Seite. Als Benannte Stelle unter der MDR auditieren wir Ihr Qualitätsmanagementsystem und führen die notwendigen Prüfungen Ihrer Technischen Dokumentationen durch.
Wir verfügen über ein globales Netzwerk, bestehend aus branchenerfahrenen Spezialisten und eigenen Laboren. Dies ermöglicht es uns, Ihnen Dienstleistungen für Medizinprodukte aus einer Hand anzubieten.
Konformitätsbewertung für Medizinprodukte und vieles mehr
Derzeit konzentrieren sich unsere Services auf die Übergangsperiode und den Zeitplan für die Umsetzung der MDR 2017/745. Unsere Experten unterstützen Sie bei der Einhaltung von Fristen. Zudem helfen sie Ihnen dabei, alle für den Zugang zu den europäischen Märkten erforderlichen Aspekte zu beachten.
Ihr vertrauensvoller Partner für den Übergang zur neuen Medizinprodukteverordnung
Als Prüf- und Zertifizierungsorganisation und Spezialist für den weltweiten Marktzugang bieten wir der Medizinprodukteindustrie umfassende Dienstleistungen aus einer Hand. Wir ermöglichen es Ihnen nicht nur, den Übergang zur neuen europäischen Medizinprodukteverordnung sicher zu vollziehen. Unsere Services umfassen darüber hinaus die Prüfung von Medizinprodukten, wie auch die Auditierung von QM-Systemen für Hersteller, Lieferanten und Vertriebsniederlassungen von Medizinprodukten. Als innovative Prüf- und Zertifizierungsorganisation sind wir auf zukünftige Themen im Bereich der Digitalisierung vorbereitet. Dazu gehören drahtlose Verbindungen, Telemedizin, medizinische Apps, Cybersecurity, der Schutz persönlicher Daten und vieles mehr.
Sprechen Sie noch heute mit einem Experten und gewinnen Sie einen Vorsprung bei der MDR-Konformität.
Fragen und Antworten zur neuen europäischen Medizinprodukteverordnung (MDR 2017/745)

Kontakt

/tuv-rheinland-de19_p05_ivd09-lp_core_4_3.jpg)
/tuv-rheinland-medical-device-single-audit-program-mdsap-ad-106934074_core_4_3.jpg)
{kind=link}