EU Medical Device Regulation MDR 2017/745

Table of contents
News: MDR transition period extended

On February 16, 2023, the European Parliament approved a proposal that will significantly ease the challenges of medical device manufacturers. With the latest extension of the Medical Devices Regulation EU 2017/745, they have the possibility to apply for an extension of the transition period for their products.
The new regulation is expected to enter into force shortly through publication in the European Journal. Manufacturers will then have the prospect of the following transition periods:
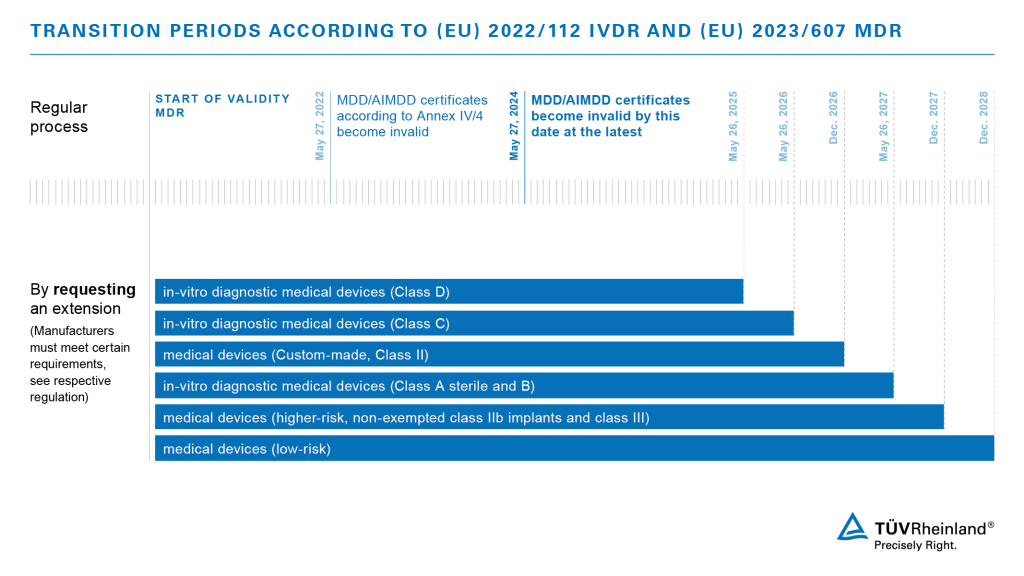
Please consider that manufacturers must demonstrate that they have already taken steps to convert to the new regulations in order to claim the extension. We offer support for all manufacturers who are looking for a professional and reliable Notified Body to address the challenges by MDR and IVDR. Contact us!
A head start on compliance with the new EU Medical Device Regulation

Manufacturers of medical devices are facing new requirements with MDR 2017/745, which took effect May 25, 2017.
The measure gives medical device manufacturers the opportunity to fully focus all possible resources on the fight against the COVID-19 pandemic. Medical device manufacturers play an important role, but challenging role. It is critically important that medical devices remain compliant and continue to be available in the EU in order to avoid shortages or delays of certain medical devices during these unique circumstances.
Beyond this situation, TÜV Rheinland would like to encourage its clients to keep in contact with us in order to continue planning activities to implement the Medical Device Regulation.
CONTACT US TO LEARN MORE ABOUT THE SPECIFICS AND TIMING OF MDR 2017/745 AND HOW IT AFFECTS YOUR BUSINESS.
MDR audits ensure access to EU markets
With the replacement by MDR 2017/745 of existing rules governing medical devices in the EU, manufacturers will soon be required to reassess their products for compliance. An MDR conformity assessment procedure provides companies the chance of achieving the necessary certification required to place products on the European market. Compliance access, your prerequisite for regulatory success.
On your path to compliance we can advise, inform and ultimately carry out any necessary technical documentation review and auditing. We are backed by a global network of industry experienced specialists and facilities and offer a one-stop shop of medical device-related services.
Medical device conformity assessment and beyond
Currently, our services focus on the transition period and the timeline for compliance with MDR 2017/745. Our experts can assist you in meeting deadlines and addressing all issues relevant to maintaining access for your medical devices to European markets.
Your confident partner for handling the replacement of the medical device directive
As a testing and certification organization and specialist for market access throughout the world, we offer the medical device industry comprehensive services all under one roof. In addition to enabling you to navigate the transition to compliance with the new European Medical Device Regulation, our services include QM system auditing for medical device manufacturers, suppliers and sales offices as well as medical device testing. We are innovative and prepared for future topics in the area of digitalization, such as wireless connections, telemedicine, medical apps, cyber security, personal data protection, and more.
Consult with an expert to get an early start on MDR compliance.
Questions and Answers on the new European Medical Devices Regulation (MDR 2017/745)
More information for download

Contact


/tuv-rheinland-de19_p05_ivd09-lp_core_4_3.jpg)
/tuv-rheinland-medical-device-single-audit-program-mdsap-ad-106934074_core_4_3.jpg)
{kind=link}