유럽(EU) 의료기기규정 MDR 2017/745

유럽(EU) 의료기기규정 MDR 2017/745
|
EU 의료기기 규정 MDR 2017/745의 적용일을 개정하는 2020년 4월 23일자 EU Regulation 2020/561이 EU 공식저널 (Official Journal)에 2020년 4월 24일 발표되었습니다. EU 의료기기규정 (EU MDR 2017/745)의 적용일은 2020년 5월 26일에서 2021년 5월 26일로 1년 유예됩니다. https://eur-lex.europa.eu/eli/reg/2020/561/oj |
|---|

TÜV 라인란드는 유럽 의료기기규정 (MDR) 2017/745 공인인증기관입니다.

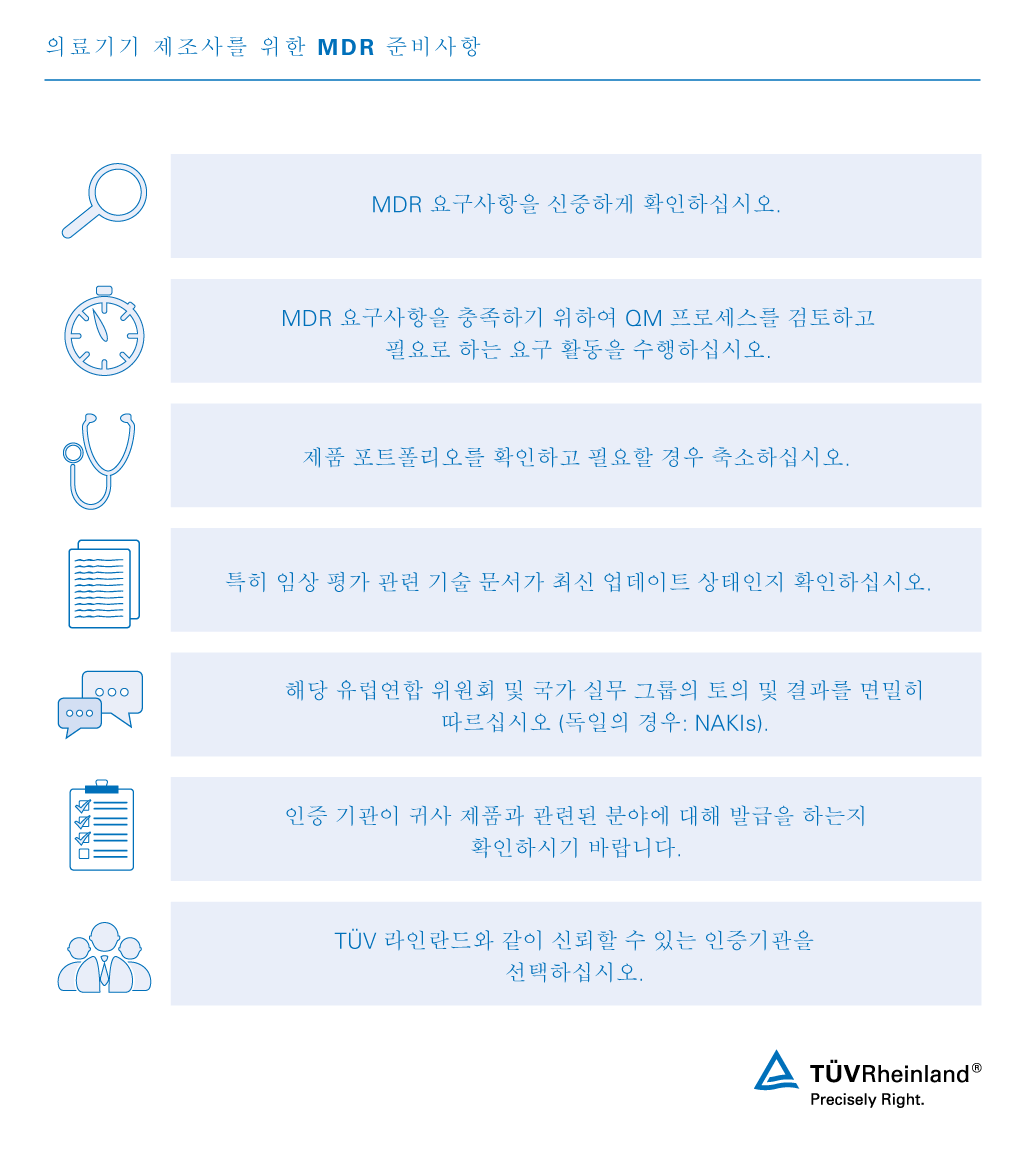
MDR 2017/745는 2017년 5월 25일에 시행되어 2021년 5월 26일자로 적용됩니다. 의료기기 제조사는 MDR 2017/745에 따른 새로운 요구사항을 준수해야 합니다.
TÜV 라인란드는 의료기기 분야에서의 견고한 신뢰성과 광범위한 전문성을 바탕으로 풍부한 실적과 경험을 보유하고 있습니다. TÜV 라인란드는 공인받은 인증기관으로서, 지금까지 활발하게 의료기기 지침 93/42/EEC (MDD) 및 능동형 이식 의료기기 지침 90/385/EEC (AIMDD)의 준수 여부를 평가해 왔으며, 이제 두 지침이 새로운 의료기기법 MDR로 대체됩니다. TÜV 라인란드는 의료기기 분야의 전문 역량과 경쟁력을 갖추고 있습니다.
현행 의료기기 법률에 따라 공인 인증기관으로서 축적한 경험과 탄탄한 위상을 바탕으로 신뢰할 수 있는 시험, 인증 파트너로서 새로운 의료기기법 MDR로 원활하게 전환할 수 있도록 지원해 드립니다.
MDR 2017/745의 세부사항 및 적용시기와 사업에 미치는 영향에 대해 자세히 알아보십시오.
MDR 심사를 통한 원활한 유럽 시장 진출
유럽연합 내의 의료기기에 적용되었던 기존 지침이 MDR 2017/745로 대체됨에 따라 제조사는 새로운 규정을 준수할 수 있도록 자사 제품을 재평가해야 합니다. MDR 적합성 평가 절차를 통해 유럽 시장에 의료기기를 판매하는 데 필요한 인증을 획득할 수 있습니다. 의료기기법 MDR 인증을 위해서는 제품이 새로운 규정의 요구사항을 충족해야 합니다.
TÜV 라인란드는 규정을 준수하여 새로운 MDR에 대비할 수 있도록 관련 정보 제공뿐만 아니라 필요한 기술 문서 검토 및 심사를 수행할 수 있습니다. 업계 경험이 풍부한 전문가 및 글로벌 네트워크를 기반으로 의료 의료 기기 관련 서비스를 원스톱으로 제공합니다.
의료기기 적합성 평가 및 기타 서비스
TÜV 라인란드는 MDR 2017/745 규정을 준수할 수 있도록 전환 기간 및 이행 계획에 대해 중점적으로 알려드립니다. TÜV 라인란드 전문가는 전환 기간 내에 새로운 유럽 의료기기 규정을 준수하여 유럽 시장에 원활하게 진출할 수 있도록 지원해 드립니다.
의료기기지침 전환을 위한 신뢰할 수 있는 파트너
TÜV 라인란드는 글로벌 시험, 인증기관으로서 해외시장 해외시장 진출에 필요한 의료기기 관련 시험, 심사, 인증 서비스를 종합적으로 제공하고 있습니다. 새롭게 시행되는 유럽 의료기기법을 준수하기 위한 전환 절차 지원뿐만 아니라, 의료기기 제조업체, 공급업체 및 영업 사무소에 대한 QM 시스템 심사를 비롯해 의료기기 시험 등 다양한 서비스를 제공합니다. 또한, 무선 연결, 원격 의료, 의료 앱, 사이버 보안,개인정보 보호 등 디지털화 분야의 미래지향적인 주제에 대한 서비스도 제공하고 있습니다.
TÜV 라인란드 전문가와 함께 MDR 규정 준수를위한 대비를 시작하십시오.
새로운 유럽 의료기기법 (MDR 2017/745)에 대한 FAQ
다운로드
| Infosheet – MDR 개정에 대한 주요 내용 | 73 KB | 다운로드 |
TUV 라인란드 의료기기 시험, 인증 서비스

150여 년 전에 설립된 TUV 라인란드는 EU 의료기기규정 MDR뿐만 아니라 체외진단의료기기법 IVDR, 의료기기 품질경영시스템 ISO 13485 인증, 의료기기 단일 심사 프로그램 MDSAP 등 글로벌 시장 진출에 필요한 의료기기 시험·인증 서비스를 원스톱으로 제공하고 있으며, 국제 표준 및 각 국가 규정에 따라 제품에 적합한 등록 절차 및 관련 교육 서비스를 제공하고 있습니다.
문의 및 견적요청


/tuv-rheinland-de19_p05_ivd09-lp_core_4_3.jpg)
/tuv-rheinland-medical-device-single-audit-program-mdsap-ad-106934074_core_4_3.jpg)
{kind=link}