EU Orvostechnikai eszköz előírás MDR 2017/745

|
Az Európai Parlament és Tanács (EU) 2020/561 szabályzata (2020. április 23.), amely módosítja a (EU) 2017/745 szabályzatot az orvostechnikai eszközökről, 2020. április 24-én jelent meg az Európai Parlament hivatalos közlönyében. A módosítás fő célja az érvényesség dátumának meghosszabbítása 2020 május 26-ról 2021 május 26-ra. Ezzel a meghosszabbítással együtt más rendelkezések érvényességi dátuma is módosult. A módosítási szabályzat hivatalos kiadványát itt tekintheti meg: https://eur-lex.europa.eu/eli/reg/2020/561/oj. |
|---|
Villámstart az új EU orvostechnikai eszköz előírás betartása terén

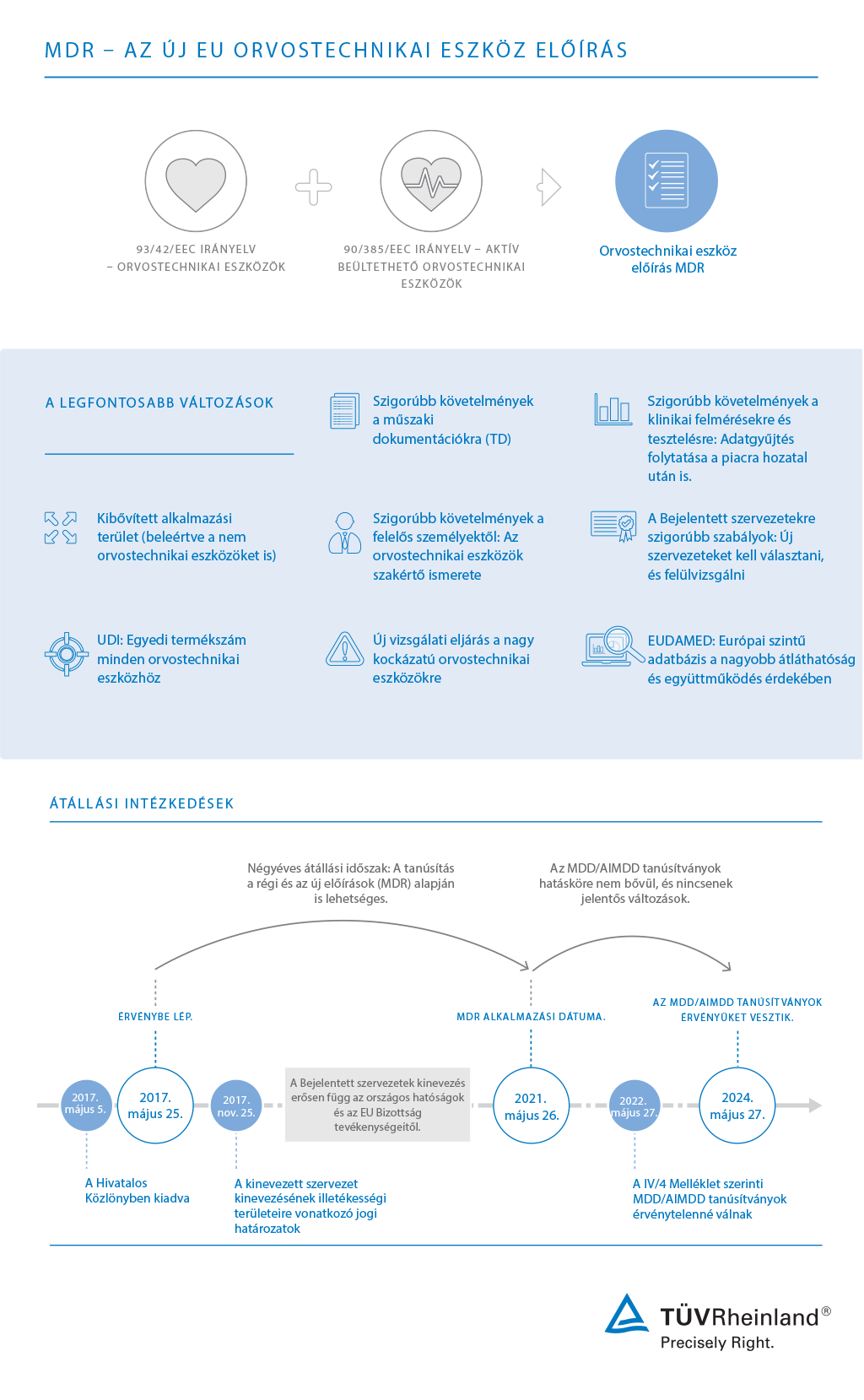
Az orvostechnikai eszközök gyártóinak új követelményekkel kell szembenéznie az MDR 2017/745 révén, amely 2017. május 25-én lép életbe. Kiadták az Európai Parlament és Tanács (EU) 2020/561 Szabályzását (2020. április 23.), amely módosítja az (EU) 2017/745 Szabályzást az Orvostechnikai eszközökről, és 2021 május 26-ra halasztja az alkalmazás dátumát. A TÜV Rheinland nagyra értékeli és támogatja a halasztást.
Ez az intézkedés lehetőséget nyújt az orvostechnikai eszközök gyártóinak, hogy minden erőforrásukat a COVID-19 járvány elleni harcra összpontosítsák. Az orvostechnikai eszközök fontos, de komoly kihívásokat jelentő szerepet játszanak. Kritikus fontosságú, hogy az orvostechnikai eszközök megfeleljenek az előírásoknak és továbbra is kaphatók legyenek az EU területén, hogy ne legyen hiány vagy késedelem bizonyos orvostechnikai eszközök esetén az ilyen egyedi helyzetek során.
A jelen helyzeten túl, a TÜV Rheinland arra buzdítja az ügyfeleit, hogy a kapcsolat fenntartásával tervezzük meg továbbra is a tevékenységeket, hogy bevezethessük az orvostechnikai eszközök előírásokat a 2021 május 26-i bevezetési dátum tekintetbe vételével.
Érdeklődjenek nálunk az MDR 2017/745 további konkrét részleteiről, és hogy ez hogyan befolyásolja az önök cégét.
MDR auditok biztosítják a bejutást az EU piacokra
Amikor az EU területén az orvostechnikai eszközökre jelenleg vonatkozó szabályokat felváltja az MDR 2017/745, a gyártóknak gyorsan újra fel kell mérnie, hogy a termékeik megfelelők-e. Az MDR megfelelés felmérésének eljárása esélyt biztosít a vállalatoknak, hogy megszerezzék a tanúsítást, amely termékeik európai piacon történő forgalmazásához szükséges. A megfelelés előfeltétele a sikeres bevezetésnek.
Miközben a megfelelés elérésén dolgoznak, javasoljuk, hogy tájékozódjanak, és hajtsák végre a műszaki dokumentációk minden szükséges módosítását és felülvizsgálatát. Az iparág tapasztalt szakértőinek és létesítményeinek világméretű hálózata támogat minket, és biztosítja számunkra az összes orvostechnikai eszközökkel kapcsolatos szolgáltatás elvégzését egy helyen.
Az orvostechnikai eszközök megfelelésének felmérése, és még több
Jelenleg a szolgáltatásaink az átállási időszakra összpontosítanak, valamint az MDR 2017/745 határidejének betartására. A szakembereink segíteni tudnak a határidők betartásában, és az összes vonatkozó kérdés megoldásában, hogy az önök orvostechnikai eszközei továbbra is bekerülhessenek az európai piacokra.
Megbízható partner az orvostechnikai eszközök irányelv szerinti átállás kezelésében
A piacokra bekerüléshez világszerte elismert vizsgálati és tanúsító szervezetként egy helyen nyújtunk minden szükséges szolgáltatást az orvostechnikai eszközök iparának. Amellett, hogy végigvisszük az új Európai orvostechnikai eszköz előírásoknak megfelelésre átállási folyamaton, a szolgáltatásaink között van a minőségirányítási rendszerek auditja orvostechnikai eszközök gyártói, beszállítói és értékesítő irodái számára, valamint az orvostechnikai eszközök vizsgálata is. Vállalatunk innovatív, és készen áll a jövőbeli kihívásokra a digitalizálás terén, mint például vezetéknélküli összeköttetés, távgyógyászat, orvosi alkalmazások, kiberbiztonság, személyi adatok védelme, és egyebek.
Konzultáljon egy szakértővel, hogy már időben elérjék az MDR megfelelést.
Kérdések és feleletek az új Európai Orvostechnikai eszköz előírásról (MDR 2017/745)
További információk letöltésre
| Tájékoztató – Fontos tények az MDR módosításáról | 73 KB | Letöltés |
Kapcsolatfelvétel

/tuv-rheinland-de19_p05_ivd09-lp_core_4_3.jpg)
/tuv-rheinland-medical-device-single-audit-program-mdsap-ad-106934074_core_4_3.jpg)

{kind=link}