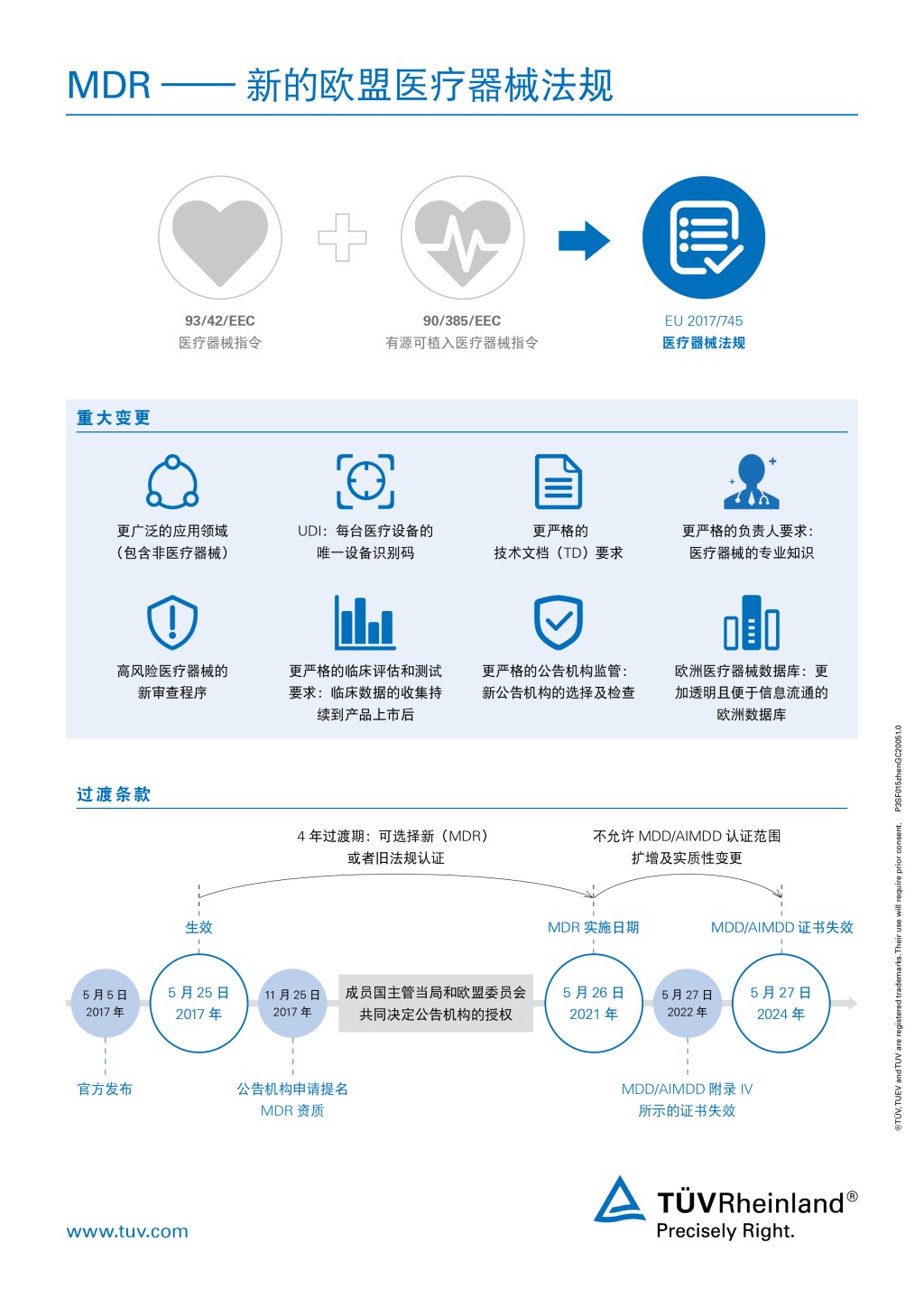
欧盟医疗器械法规 MDR 2017/745

|
2020年4月24日,《欧洲议会官方公报》发布(EU) 2020/561法规,对医疗器械法规(MDR) [REGULATION (EU) 2017/745]进行了修订,将MDR的执行日期从2020年5月26日推迟到2021年5月26日,并对其他规定的执行日期也做了相应调整。有关《修订条例》的详细内容请点击: https://eur-lex.europa.eu/eli/reg/2020/561/oj |
|---|
在新的欧盟医疗器械法规的符合性方面领先一步

医疗器械制造商正面临MDR 2017/745的新要求,该法规于2017年5月25日生效,强制实施日期为2021年5月26日。
医疗器械制造商在抗击COVID-19疫情中扮演着重要角色,推迟执行MDR将有助于医疗器械保持合规性,以便继续在欧盟可用,避免在特殊情况下某些医疗器械的短缺或延误。目前,TÜV莱茵正在集中资源配合企业完成COVID-19疫情相关产品MDD/MDR法规的 CE认证,保证相应医疗器械产品能尽快合规地出口市场。
除此之外,TÜV莱茵将持续关注和积极应对MDR延迟带来的变化。作为当前医疗器械法规的公告机构,我们拥有丰富的经验和稳固的地位,能够全力支持您过渡到新法规。
请联系我们,了解更多关于MDR 2017/745的细节和时间安排,以及它如何影响您的业务。
MDR审核确保客户进入欧盟市场
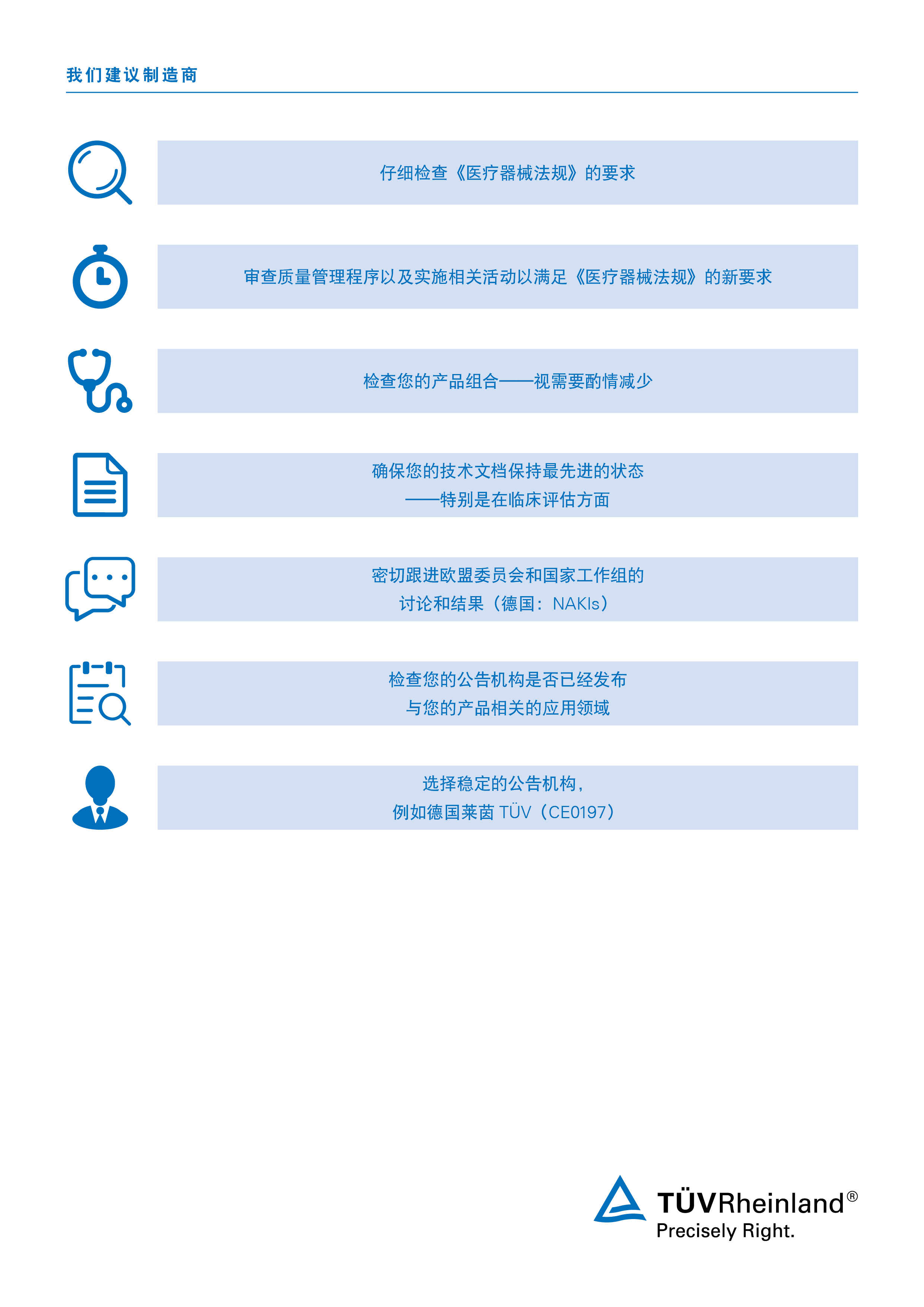
随着欧盟医疗器械的现行规则被MDR 2017/745取代,制造商将很快被要求重新评估其产品的合规性。MDR符合性评估程序帮助公司获得其将产品投放欧洲市场所需的必要认证。合规进入,是审批成功的先决条件。
在您的合规审批之路上,我们可以提供建议、通知并最终执行任何必要的技术文档评审和审核。我们有一个由经验丰富的行业专家和设施组成的全球网络作为后盾,并提供一站式医疗器械相关服务。
医疗器械符合性评估及其他
目前,我们的服务侧重于过渡期和遵守MDR 2017/745的时间表。我们的专家可以帮助您在截止日期前完成工作,并解决与保证医疗器械进入欧洲市场相关的所有问题。
您处理替换医疗器械指令的坚实合作伙伴
作为全球 市场准入 的测试和认证组织及专家,我们团队为医疗器械行业提供全面的服务。除了使您能够顺利过渡符合新的欧盟医疗器械法规,我们的服务还包括医疗器械制造商、供应商和销售办事处的质量管理体系审核以及医疗器械测试。我们具有创新精神,并为未来数字化领域的主题做好了准备,如无线连接、远程医疗、医疗应用、网络安全 、个人数据保护等。
请咨询专家,以尽早开始着手MDR符合性审核 。
關於新的歐盟醫療器材法規(MDR 2017/745)的問答
| 信息表 – MDR修订版的重要内容 | 73 KB | 下载 |
更多信息下载
联系我们


/tuv-rheinland-medical-device-single-audit-program-mdsap-ad-106934074_core_4_3.jpg)
{kind=link}