In Vitro Diagnostic Medical Device Regulation (IVDR)
/tuv-rheinland-de19_p05_ivd09-lp_core_2_2_1.jpg)
Table of contents
TÜV Rheinland LGA Products GmbH is a EU designated Notified Body under the In Vitro Diagnostic Device Regulation (IVDR 2017/746)
We are pleased to announce, that TÜV Rheinland is officially a desginated Notified Body under In Vitro Diagnostics (IVDR 2017/746), and accepting conformity assessment applications from 28th of November 2020.
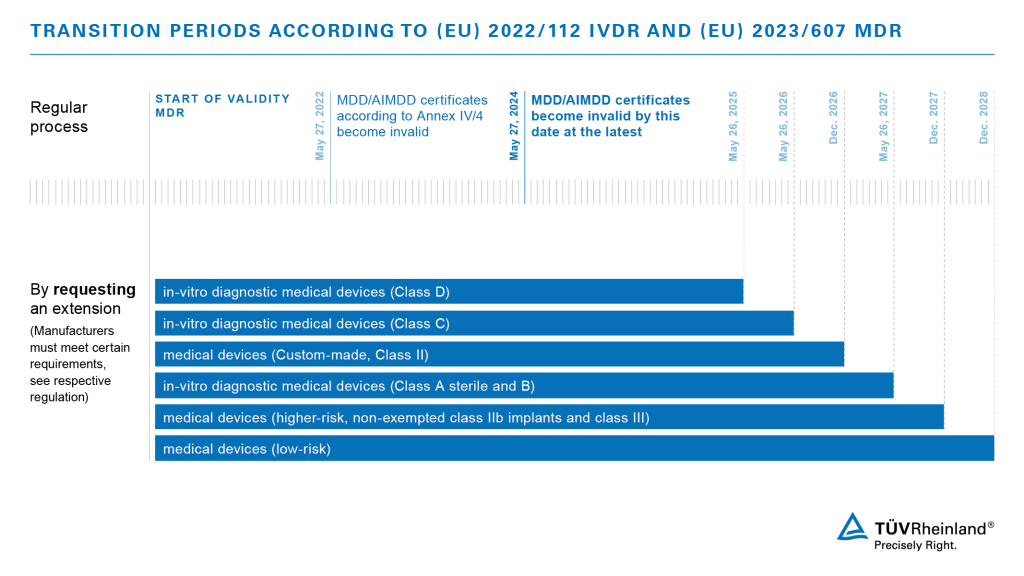
Approach IVDR compliance with confidence
/tuv-rheinland-ivdr-visual-1-en_core_1_x.png "Important changes from IVDD to the new IVDR")
Respected manufacturers of in vitro diagnostic devices value certification as a way to demonstrate their commitment to the highest standards of quality, meet European market requirements and adorn the CE marking.
Manufacturers are facing new requirements under IVDR 2017/746 issued by the European Commission (CE), which include more stringent mandates such as increased accountability, transparency, detailed documentation and regular filings to the new EUDAMED database.
The most significant change is the new product classification system, and the requirement that manufacturers of IVD devices (Class A exempted) must consult a Notified Body for approval.
Successful transition to IVDR compliance requires manufacturers to prepare for reliable documentation of performance, high safety standards and effective risk management, in addition to conducting comprehensive product inspections while on (and prior to entering) the EU markets.
We provide comprehensive services for your initial or transitionary compliance with the new In Vitro Diagnostic Devices Regulation. Our full range of medical testing and certification services enables us to offer you a tailored service suited to your products and needs.
Our application for designation under regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) is pending, and expected to be approved by December 2020.
CONTACT US TO LEARN MORE ABOUT THE SPECIFICS AND TIMING OF IVDR 2017/746, AND HOW IT AFFECTS YOUR BUSINESS.
Preparatory recommendations for the IVDR compliance process
Pre-compliance checks (general recommendation):
- Classify products
- Establish working groups to setup UDI numeration strategy
- Perform gap analysis
- Close gaps in QMS (special focus on post-market surveillance & risk management)
- Close gaps in technical documentation (special focus on performance evaluation & clinical evidence)
- Coordinate with the Notified Body
Our services include auditing, assessment, testing, inspection, and issuance of regulatory documentation. We inform you on issues related to transition deadlines relevant to certification and addressing issues related to maintaining uninterrupted compliance status and European market access for your devices.
Gain access to European markets with IVDR compliant products
Securing your product compliance with the new EU regulation is mandatory for CE marking and entry into European markets. CE mark certificates are issued based on a successful conformity assessment, and shows your commitment to excellence and fulfillment of European market requirements to authorities and customers.
The CE mark on your in vitro diagnostic device can also increase your competitiveness on the global market beyond EU nations. Our customers enjoy a single-source provider experience from around the world, and have access our network of highly qualified medical device experts sitting locally in major markets.
Your globally recognized and trusted IVDR partner
Our global network of regulatory experts excel in conducting assessments according to the new IVDR requirements. Our reputation for excellence reflects our near 150 years of experience serving a diverse range of industries and customers.
We pride ourselves in delivering high-quality tailored services from a single source to save you time, money and provide the evaluations you need to succeed in the competitive international medical device market.
Complete the IVDR compliance process and maximize market access opportunities with confidence through a single source provider.
FAQ’s on IVDR
Questions and Answers on the new European In Vitro Diagnostic Medical Devices Regulation (MDR 2017/746)
More information on IVDR
| Brochure: Product testing and certification for medical devices | 3 MB | Download |
Related Topics
Contact



_core_4_3.jpg)
/tuv-rheinland-ivdr-visual-1-en.png){kind=link}